Tutorial¶
Technical background¶
20/20+ uses the random forest algorithm to predict cancer driver genes. Because 20/20+ uses supervised machine learning, cancer driver gene prediction depends on a list of defined oncogenes and tumor suppressor genes used for training. The list is already pre-defined from the Cancer Genome Landscapes paper. Included in the results are a score specifically for oncogene or tumor suppressor gene, and a cumulative driver gene score. The score represents the fraction of decision trees in the random forest which voted for the particular class (oncogene, TSG, driver (either oncogene or TSG), passenger). Scores nearer one indicate stronger evidence for the gene as a cancer driver. There are two ways to use 20/20+ scores. One is to just obtain cancer driver scores for genes, which allows ranking of genes in a prioritized manner. The second way evaluates the statistical significance of the cancer driver score. A p-value and associated Benjamini-Hochberg false discovery rate will be reported, but this requires establishing the null distribution for random forest scores. 20/20+ uses an empirical null distribution by scoring simulated mutations in genes. This extra step requires additional work and computational resources. 20/20+ can be applied to pan-cancer and tumor type specific data. 10-fold cross-validation is performed internally within 20/20+ to avoid overfitting.
20/20+ pipeline¶
The easiest way to run the entire 20/20+ pipeline from somatic mutations to cancer driver gene prediction is to use snakemake. We have created a Snakefile that will run the multiple steps needed to get the final results. You first will need to install snakemake (requires python 3.X), so please see the snakemake installation instructions. You should be able to see the snakemake help, if you are in the 2020plus directory and it is installed correctly.
$ snakemake --help # help using snakemake
$ snakemake -q help # help for 2020plus commands
There are two ways to perform predictions with 20/20+. Either in a pan-cancer setting where mutations from several cancer types are aggregated together, or predicting cancer type specific driver genes by using a 20/20+ model previously trained on pan-cancer data.
Cancer type specific analysis¶
Preparing data¶
To set up this tutorial example you will first need to download the data. This step is needed regardless of whether your doing predictions on pan-cancer or cancer type specific data.
$ mkdir data
$ cd data
$ wget https://www.dropbox.com/scl/fi/qdlwws3u4s317cfvxf4f2/bladder.txt.gz?rlkey=e7ihy09c7nrbqib5cnn26pne7&st=9vr4py97&dl=1 -o bladder.txt.gz # download mutations
$ wget https://www.dropbox.com/scl/fi/jnucugcu4qslb8vw0s9ry/snvboxGenes.bed?rlkey=yc3gqu4msx0wgqb6wo149rpok&st=ppij68wa&dl=1 -o snvboxGenes.bed # download transcript annotation
$ wget https://www.dropbox.com/scl/fi/o6eaih9d3rr9ztms2pa33/scores.tar.gz?rlkey=ekih9qstzfncn8811935a7ghb&st=58ife2e0&dl=1 -o scores.tar.gz # download pre-computed scores
$ wget https://www.dropbox.com/scl/fi/zv3twjeii2ghxtgy4f9o5/2020plus_10k.Rdata?rlkey=yu8i09tuuf6bbgfzsm7dcynp8&st=usug1aew&dl=1 -o 2020plus_10k.Rdata # download pre-computed scores
$ gunzip bladder.txt.gz
$ tar xvzf scores.tar.gz
$ cd ..
The somatic mutations in the mutations.txt file is a MAF-like format described here. snvboxGenes.bed contains reference transcripts for each gene (described here), and the scores directory contains pre-computed scores (described here). You will also need to create a FASTA file of gene sequences by the following these instructions.
Running 20/20+¶
When performing predictions on cancer type specific mutations, a pre-trained 20/20+ classifier based on pan-cancer data is used to make predictions. Available pre-trained 20/20+ classifiers are shown on the Download page. Associated data should be collected like in the above Preparing data section. When using a pre-trained classifier, the snakemake -s Snakefile pretrained_predict command should be used. In the below example command, we use the command for a local machine, but it can be adopted to run on a cluster.
$ snakemake -s Snakefile pretrained_predict -p --cores 1 \
--config mutations="data/bladder.txt" output_dir="output_bladder" trained_classifier="data/2020plus_10k.Rdata"
The –cores argument specifies the number of computer cores that are allowable to be used at a given time. In this example, the output will be saved in the “output_bladder” directory as specified by the output_dir parameter (also changeable in config.yaml).
It is generally recommended to run 20/20+ on a cluster to parallelize calculations. The below command will execute the 20/20+ pipeline on an SGE computer cluster using qsub. The cluster submission command can be changed to fit your particular cluster scheduler.
$ snakemake -s Snakefile pretrained_predict -p -j 999 -w 10 --max-jobs-per-second 1 \
--config mutations="data/bladder.txt" output_dir="output_bladder" trained_classifier="data/2020plus_10k.Rdata" \
--cluster-config cluster.yaml \
--cluster "qsub -cwd -pe smp {threads} -l mem_free={cluster.mem},h_vmem={cluster.vmem} -v PATH=$PATH"
The –cluster argument specifies the command prefix for submitting to your cluster job scheduler. In the above example, qsub is used for the SGE scheduler, but this obviously is cluster specific and therefore you should look up the manual for your cluster. Of importance, though, is that certain template values can be inserted in to the job submission. Templated values are denoted by curly braces, and are used to set the number of threads (“{threads}”) and memory (“{cluster.mem}” and “{cluster.vmem}”). Templated values with “cluster.” are specified in the cluster config file (cluster.yaml; –cluster-config argument). It is also recommended that your PATH environmental variable is passed into the cluster job submission so that you do not receive a command not found error. The “-j” argument can restrict the number of concurrent jobs submitted to the cluster,but in our case we use 999 to let the cluster job scheduler to identify which jobs get executed. The “-w 10 –max-jobs-per-second 1” parameters are issued to avoid overly quick job submissions to the cluster. The difference with the next pan-cancer tutorial is that the mutations (“data/bladder.txt”) are from a single cancer type, and the pre-trained classifier is specified with the trained_classifier option. In this case the pre-trained 20/20+ classifier was assumed to be placed into the data directory.
Note
The run time of 20/20+ depends on the number of simulations. By default, the NUMSIMULATIONS is set at 10000, which is lower than used in the original 20/20+ paper. This can be increased via the NUMSIMULATIONS variable (e.g. from 10000 to 100000) in the config.yaml file or specification in the command line of snakemake via –config NUMSIMULATIONS=100000. This might result in a slight increase in prediction performance but may be too time consuming for large data. Make sure you use the correct trained classifier based on your NUMSIMULATIONS option, by using 2020plus_100k.Rdata for NUMSIMULATIONS=100000 and 2020plus_10k.Rdata for NUMSIMULATIONS=10000.
20/20+ output¶
Like in the quick start, you will find the result in output_bladder/results/r_random_forest_prediction.txt. There will be a p-value/q-value for the oncogene, tumor suppressor gene, and driver score. At a false discovery rate of 0.1, you should get 9 significant oncogene scores, 34 significant TSG scores, and 46 significant driver scores. The file will also contain all of the features used for prediction. Examine the QQ plot of p-values as a diagnostic check on the reported p-values (output_bladder/plots/qq_plot.png). You will need the matplotlib python package installed for the plot to be created (see installation instructions). The observed p-values (blue line) should be close to the theoretically expected p-values (red line). In this case, the mean absolute log2 fold change (MLFC) indicates that the p-values are in good agreement with expectations. Please see our paper for more discussion on the MLFC.
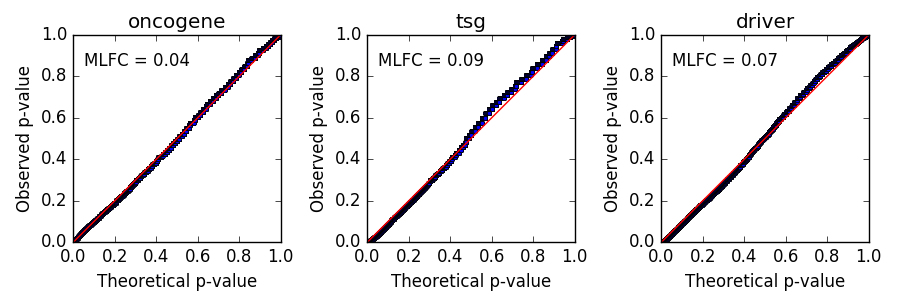
A mean absolute log2 fold change (MLFC) of greater than 0.3 may indicate problems with the null distribution. One cause of high MLFC are problems in the provided mutations. For example, if mutations of both a primary tumor and metastasis were provided or data arising from low quality mutation calls (e.g. caused by read mapability problems, etc.). Further quality control of the mutation data could fix the problem. In scenarios where mutation quality does not appear to be the cause, a more stringent false discovery rate threshold may be needed, or just rely on the random forest score without placing emphasis on the reported p-value.
Pan-cancer analysis¶
Note
The pan-cancer tutorial is more computationally intensive and the run time will take a while even on a computer cluster. However, it does demonstrate the correct usage of the 20/20+ pipeline, which is quicker for cancer type specific data sets.
The snakemake -s Snakefile predict command will perform predictions on pan-cancer data. Here, it is assumed you are in the 2020plus directory where the Snakefile is located.
Preparing data¶
To set up this tutorial example you will first need to download the data. This step is needed regardless of whether your doing predictions on pan-cancer or cancer type specific data.
$ mkdir data
$ cd data
$ wget https://www.dropbox.com/scl/fi/8ob367fu9ztplyx4mmcj0/pancan-mutation-set-from-Tokheim-2016.txt.gz?rlkey=nxxwkotnuggw2ptinjbbfvp96&st=lw6ah2ip&dl=1 -o pancan-mutation-set-from-Tokheim-2016.txt.gz # download mutations
$ wget https://www.dropbox.com/scl/fi/jnucugcu4qslb8vw0s9ry/snvboxGenes.bed?rlkey=yc3gqu4msx0wgqb6wo149rpok&st=ppij68wa&dl=1 -o snvboxGenes.bed # download transcript annotation
$ wget https://www.dropbox.com/scl/fi/o6eaih9d3rr9ztms2pa33/scores.tar.gz?rlkey=ekih9qstzfncn8811935a7ghb&st=58ife2e0&dl=1 -o scores.tar.gz # download pre-computed scores
$ gunzip pancan-mutation-set-from-Tokheim-2016.txt.gz
$ mv pancan-mutation-set-from-Tokheim-2016.txt mutations.txt # rename file
$ tar xvzf scores.tar.gz
$ cd ..
The somatic mutations in the mutations.txt file is a MAF-like format described here. snvboxGenes.bed contains reference transcripts for each gene (described here), and the scores directory contains pre-computed scores (described here). You will also need to create a FASTA file of gene sequences by the following these instructions.
Running 20/20+¶
By default, the data is assumed to be located in the “data/” directory and mutations are “data/mutations.txt”. You can change the default by editing the config.yaml file. However you can also override the default from the command line by specifying variables with the –config argument. The following command executes the 20/20+ on a local machine.
$ snakemake -s Snakefile predict -p --cores 1 \
--config mutations="data/mutations.txt" output_dir="output_pancan"
The –cores argument specifies the number of computer cores that are allowable to be used at a given time. In this example, the output will be saved in the “output_pancan” directory as specified by the output_dir parameter (also changeable in config.yaml).
It is generally recommended to run 20/20+ on a cluster to parallelize calculations. The below command will execute the 20/20+ pipeline on an SGE computer cluster using qsub, like in the previous cancer type specific analysis. The cluster submission command can be changed to fit your particular cluster scheduler.
$ snakemake -s Snakefile predict -p -j 999 -w 10 --max-jobs-per-second 1 \
--config mutations="data/mutations.txt" output_dir="output_pancan" \
--cluster-config cluster.yaml \
--cluster "qsub -cwd -pe smp {threads} -l mem_free={cluster.mem},h_vmem={cluster.vmem} -v PATH=$PATH"
20/20+ output¶
Like in the quick start, you will find the result in output/results/r_random_forest_prediction.txt. There will be a p-value/q-value for the oncogene, tumor suppressor gene, and driver score. The file will also contain all of the features used for prediction.
Train a 20/20+ classifier¶
You can also train your own 20/20+ model to predict on new data (e.g. new cancer type specific data) using the train command. Training should be performed on a pan-cancer collection of mutations. This either could be those mutations used in our evaluation or a new collected set. Note, the provided pre-trained classifier on the downloads page is already trained on the mutations linked in the previous sentence. The file format for mutations is described here. Like above, the command can be easily modified to run on a cluster.
$ snakemake -s Snakefile train -p --cores 1 \
--config mutations="data/my_pancancer_mutations.txt" output_dir="output"
where “data/my_pancancer_mutations.txt” is the file containing small somatic mutations and the trained 20/20+ model will be saved as “output/2020plus.Rdata”.